More Detail on Peri-Substitution Research
To expand the scope of what is possible in terms of bonding, structure and reactivity is central to modern main group chemistry. For example, development of base stabilisation allowed synthesis of new low coordinate p-block species, which can be considered as new allotropes of p-block elements with fundamentally differing properties, such as good solubility in organic solvents.
Our major interests include stabilization of a variety of normally fleeting Group 15 environments and bonding situations utilizing the special geometric features of peri-substitution. Peri-substitution is substitution in positions 1 and 8 in naphthalene or related polyaromates such as acenaphthene and anthracene. The uniqueness of the peri-geometry is demonstrated in the graphical representation of peri-, ortho– and bay-region geometries (Figure 1, top). Although all these backbones are rigid and planar, they differ in the distance to which they constrain the two substituents (Figure 1). Peri-substitution places the peri-atoms into much closer proximity than ortho–substitution. This results in the change of the preferred way of achieving a relaxed geometry, which in case of peri-substitution is formation of a direct bond between the two peri-atoms (Figure 1).
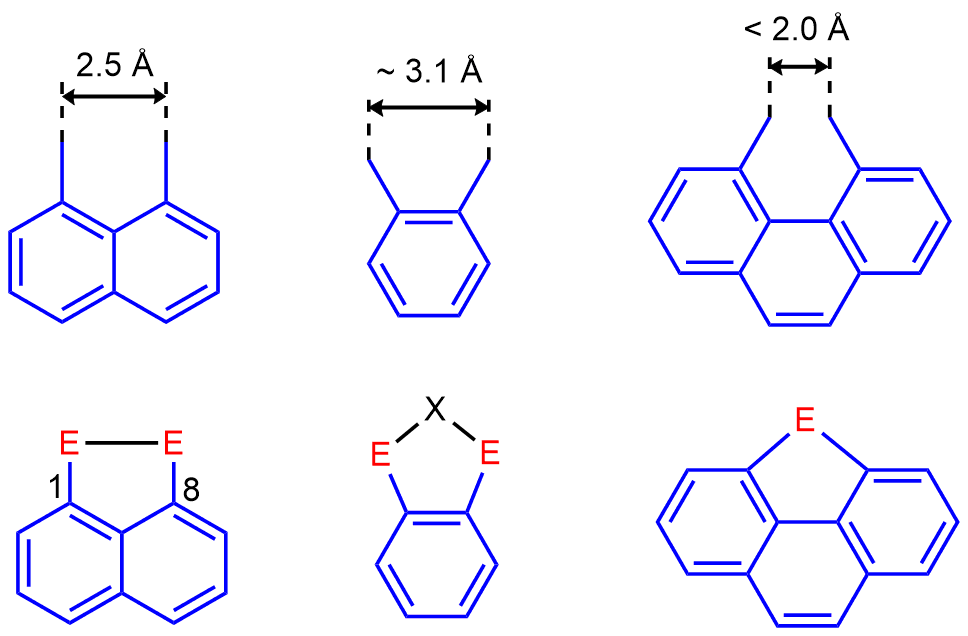
Figure 1. Comparison of the geometry of peri-substitution in naphthalene, ortho-substitution in benzene, and bay-region disubstitution in phenanthrene. The preferred way of achieving a relaxed geometry for each motif is shown at the bottom.
Peri-substitution is special in two ways. Firstly, by attaching substituents into peri-positions via strong and inert P-C bonds, the two substituents become locked in one molecule and therefore cannot drift apart and do their own chemistry as two separate entities. Secondly, the rigid polyaromate acts as a clamping framework, in which the peri-substituents are forced into close proximity (ideally to a distance of approx. 2.5 Å from each other). Placing the peri-substituents at this distance is associated with relaxed (i.e. planar) geometry of the backbone, free from angular strain. Therefore, the bonding interaction between peri-atoms is highly favoured, since the resulting P-P bond distance (usually ≈ 2.2 Å) is rather close to the natural peri-distance. Repulsive interaction between the two peri-functionalities is normally much longer than 2.5 Å and therefore it is less favourable. In such situation the two peri-atoms are forced together by the organic backbone, but this leads to sub-van der Waals contacts. A compromise structure is achieved, featuring some sub-van der Waals contacts as well as distortions of the organic backbone (angular strain) (see Figure 2). Even with this compromise, the energy stored in the system (similar to that of compressed spring) is substantial. It is this strain energy difference between the bonding and non-bonding that results in the unusual reactivity, structure and bonding in peri-substituted motifs.
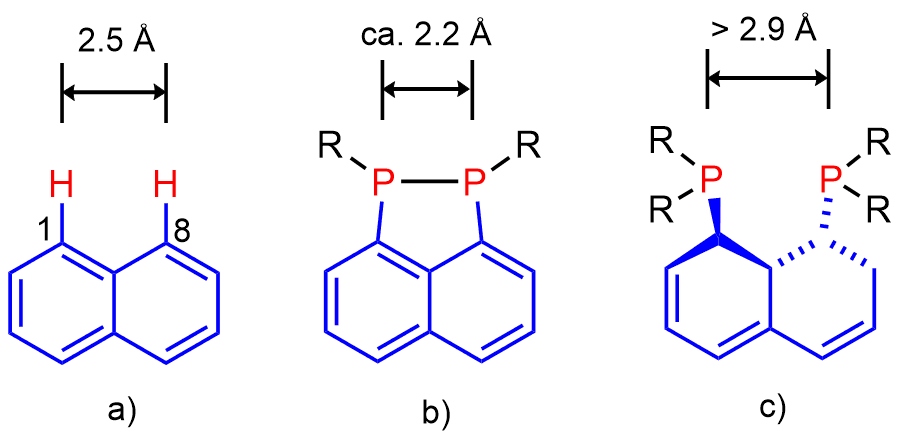
Figure 2. a) Natural distance of peri-atoms in naphthalene. All bond angles in naphthalene are 120º, therefore the two bay region C-H bonds are parallel. Metric parameters of naphthalene result in natural distance of peri-atoms being ca. 2.5Ǻ. b) Example of (favoured) bonding interaction between peri-atoms. The two exo C-P bonds are close to parallel in this situation. c) Example of repulsive interaction between peri-atoms, resulting in significant amount of angular strain (which may include twisting of the naphthalene backbone) and sub-van der Waals contacts.
The strong preference for bonding geometry and resulting unusual reactivity is shown in the example in Figure 3. Mild hydrolysis of aryldichlorophosphines, such as PhPCl2, proceeds cleanly affording a single product – phosphinic acid (Figure 3, top). No redox change occurs during the reaction; both the starting material and the product contain phosphorus in oxidation state III.
In contrast, hydrolysis of the naphthalene derivative 1,8-C10H6(PCl2)2 (Figure 3, bottom) is much more complex than one would expect based on the above example. Two products are formed, both having P-P bonded geometry. The formal oxidation states of phosphorus atoms in these products are I, III, IV and V.[1]

Figure 3. Hydrolysis of prototypical aryldichlorophosphine (top) and related peri-naphthalene dichlorophosphine (bottom).
As indicated earlier, we are using peri-substitution to generate unusual bonding, in particular to stabilise normally fleeting structural motifs. For example, using peri-substitution in acenaphthene, we have been able to stabilize a phosphine-phosphine donor-acceptor adduct shown in Figure 4.[2] The adduct is the first compound of this type stable at room temperature, all other known examples decompose well below 0°C. Its “bottleability” allowed us to accomplish the first systematic reactivity study of a compound from this class.
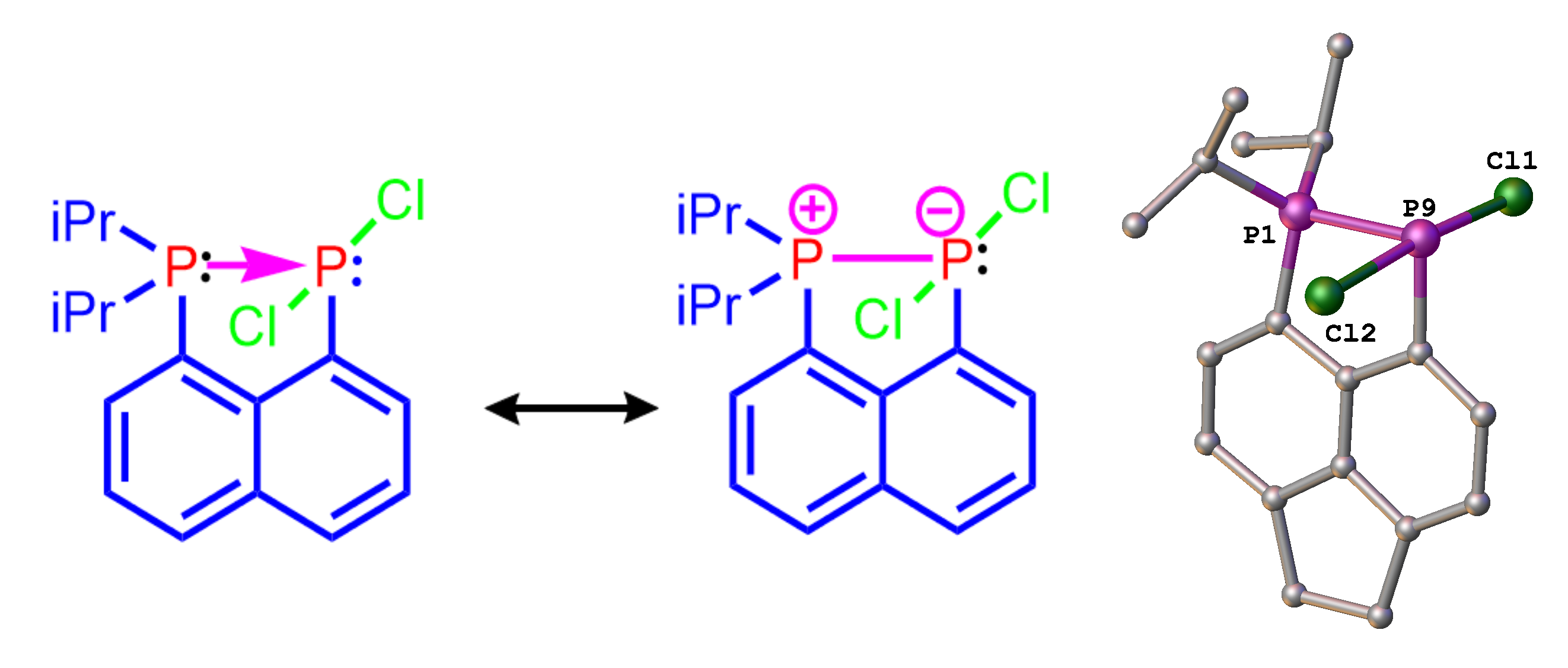
Figure 4. Mesomeric structures (left and middle) and X-ray structure (right) of phosphine→phosphine donor, stabilized using the peri-geometry.
As mentioned recently by Protasiewicz,[3] whilst of fundamental interest, no bis(borane) push-double pull phosphinidene systems have been structurally characterized previously. We have prepared and fully characterised such a bis(borane) complex (see Figure 5) via the reaction of our phosphine-phosphine adduct with borane dimethylsulfide.[4] Despite literature mentioning several attempts to make compounds of this class, only very few were successful. Because of this, one would expect these adducts to be very reactive and/or highly thermally unstable species. However, our bis(borane) adduct turns out to be indefinitely stable at room temperature, and even more intriguingly, it can be manipulated in air as it is insensitive to oxygen or moisture. The X-ray diffraction confirmed the bis(borane) displays an interesting bonding situation in which a Lewis base-stabilized phosphinidene moiety acts as a double donor towards two Lewis-acidic (borane) moieties, i.e. push-double pull arrangement. Two representative resonance forms (zwitterionic and with DA bonding) are shown in Figure 5.
Figure 5. Mesomeric structures (left) and X-ray structure (right) of the push-double pull bis(borane) complex.
References
[1] P. Kilian, H. L. Milton, A. M. Z. Slawin, J. D. Woollins, Inorg. Chem. 2004, 43, 2252-2260. DOI: 10.1021/ic035364h
[2] P. Wawrzyniak, A. L. Fuller, A. M. Z. Slawin, P. Kilian, Inorg. Chem. 2009, 48, 2500-2506. DOI: 10.1021/ic801833a
[3] D. V. Partyka, M. P. Washington, J. B. Updegraff III, R. A. Woloszynek, J. D. Protasiewicz, Angew. Chem. Int. Ed. 2008, 47, 7489 – 7492.
[4] B. A. Surgenor, M. Bühl, A. M. Z. Slawin, J. D. Woollins, P. Kilian, Angew. Chem. Int. Ed. 2012, 51, 10150 –10153. DOI: 10.1002/anie.201204998 OPEN ACCESS